Solving crystal structure of GPCR-G protein complex
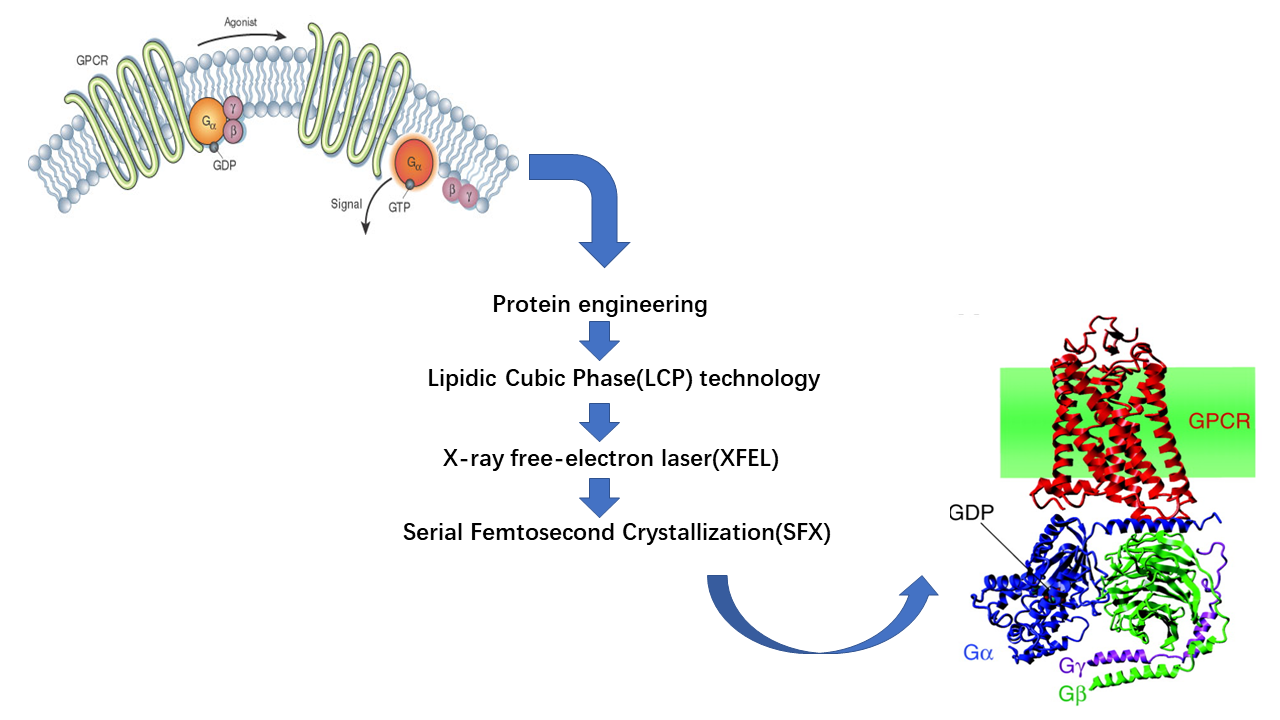
GPCR constitutes 30%-40% drug targets in current pharmaceutical industry, however, the lack of high-resolution structure largely hinder the further investigation to harness their desirable potential. As a membrane protein, GPCR is conformationally flexible and dynamic. A plethora of approaches has been proposed to resolve this issue, including protein engineering(site-directed mutation, fusion protein, protein truncation). Our lab employs the state-of-art lipidic cubic phase(LCP) technology to specifically assist in GPCR protein crystallization. Due to its amphiphilic property which perfectly mimicking natural phospholipid bilayer, LCP enables the hydrophobic and hydrophilic fragment of GPCR protein diffuse freely within the system and more amenable to form lattice. Followed by X-ray free-electron laser(XFEL) and Serial femtosecond crystallization(SFX) technology, which make it possible to generate high-quality data from microcrystals. We have elucidated dozens of GPCR proteins’ structure and their underlying regulatatory mechanisms.
In my project(also my undergraduate thesis), I try to optimize current pipelines for construct designing, GPCR protein extracting and to explore the novel approaches to stablize GPCR protein. To be specific, we adopt the concept of solving GPCR-G protein complex instead of merely solving GPCR structure. There are there reasons for that:1) GPCR protein is highly dynamic and flexible, G protein could serve as a stablizer to immobilize GPCR protein and force them retaining natural and functional state. 2) By using G protein to form the complex, we are able to increase the overall molecular weight of GPCR protein, which may offer us a new method to solve its protein through Cryo-EM. 3) We hope to illustrate how GPCR interact with downstream signal partner and further extend our understanding.
Using Machine-learning approach to streamline trial-and-error process during sample preparation
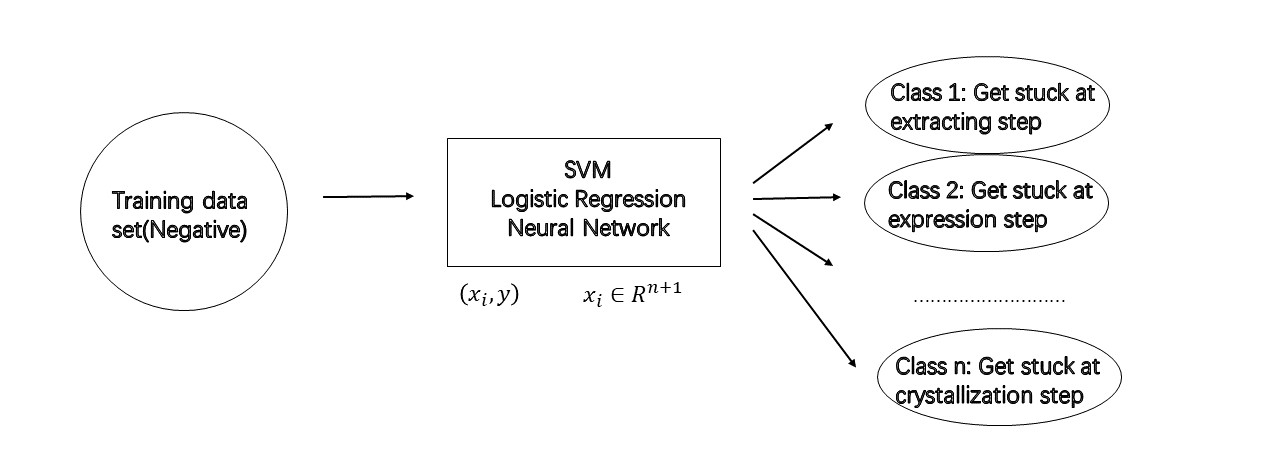
In order to simplify and streamline the time and labor consuming process during sample preparation, I endeavored to modify existing machine learning algorithm and apply them into predicting the effect of particular protein engineering approaches, including fusion protein, site-directed mutaton and protein truncation. The idea here is to adopt multi-classfication instead of current binary classifier. It is exceedingly benefit, when we hope to carry on one kind of engineeing approaches(Say mutate site K6.43), binary classfier could only tell you if it could work, if not, the only thing left we can do is to arbitrarily find another site. In contrast, a multi-classification could let us know which step could be exactly most likely getting stuck, then we are able to apply our biochemistry knowledge to do subsequent analysis and derive another possible site with higher confidence.
By doing that, firstly we need to collect training data set as mush as possible. Fortunately, there are tons of negative data generated from our lab(Which is imaginable, optimizing protocol is a high-failure rate process). We assign them a series of score from 0 to n corresponding to the certain stip upon which they would get stuck. Next we try to find feature vectors as following:
Sequence-based features (Hydrophobicity, Hydrophilicity, Amino acid percentiles .. )
Structure-based features (Van del Waals Force, Salt bridge, Hydrogen bond ..)
Energy-based features (Gibbs free energy ..)
Then we fit those parameter depending on which machine learning algorithm we chose and finally it may work!
Reference
- Vilardaga, Jean-Pierre, et al. “G-protein-coupled receptor heteromer dynamics.” J Cell Sci 123.24 (2010): 4215-4220.
- Li, J. et al. The Molecule Pages database. Nature 420, 716-717 (2002).
- Popov, Petr, et al. “Computational design of thermostabilizing point mutations for G protein-coupled receptors.” eLife 7 (2018): e34729.